Protein Trafficking and Neurodegenerative Diseases
INITIATION DATE:
12.01.09
ARC DIRECTORS AND CO-DIRECTORS:
Lindsay Farrer, PhD; Director; Professor & Chief; Medicine/ Neurology/ Genetics & Genomics/ Epidemiology/ Biostatistics
OVERVIEW OF GOALS AND MISSION:
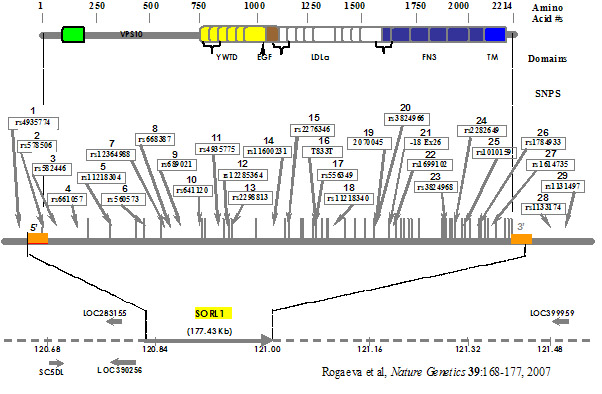
Recycling of the amyloid precursor protein (APP) from the cell surface via the endocytic pathways plays a key role in the generation of amyloid ß-peptide (Aß), the accumulation of which is thought central to the pathogenesis of Alzheimer disease (AD). This proposed ARC is the outgrowth of our major discovery of SORL1 as contributing to AD susceptibility (Nature Genetics 2007; 39:168-177). We observed association of several haplotypes in two distinct regions of SORL1 in seven diverse populations and showed that these variants may regulate tissue-specific expression of SORL1, which directs trafficking of APP into recycling pathways. Our work also showed that these variants may regulate tissue-specific expression of SORL1, which directs trafficking of APP into recycling pathways. When SORL1 is under-expressed, APP is sorted into Aß-generating compartments. More importantly, this body of evidence firmly establishes abnormalities and perhaps even normal variation in protein trafficking as a fundamental provocateur in AD pathogenesis. These mechanisms may involve a diverse group which regulate sorting, recycling, sequestration and metabolism of APP, other proteins which interact with APP, or intracellular structures which influence the synthesis or trafficking of APP (e.g., golgi, endoplasmic reticulum, mitochondria). This ARC will explore the role of vesicular sorting proteins and other genes involved in protein trafficking in the etiology and pathophysiology of AD and other neurodegenerative disorders. The power of this ARC lies in its ability to validate any finding using independent approaches of genetic epidemiology, cell biology, model systems and pathology. To achieve this goal, we assembled a nucleus of scientists from multiple disciplines on both campuses.
ARC Members:
| Faculty Name / Degree | Academic Rank | Department(s) |
| Lindsay Farrer, Ph.D. (Director) * | Professor & Chief | Medicine, Neurology, Ophthalmology, Epidemiology, Biostatistics |
| Carmela Abraham, Ph.D. * | Professor | Biochemistry, Medicine |
| Uwe Beffert, Ph.D. | Res. Assistant Professor | Biology |
| Victoria Bolatina, Ph.D. | Professor | Medicine |
| Clinton Baldwin, Ph.D. * | Adjunct Professor | Medicine |
| Ci-Di Chen, Ph.D. * | Research Assistant Professor | Biochemistry |
| Libin Cui, Ph.D. | Research Assistant Professor | Pharmacology |
| Richard Fine, Ph.D. | Professor | Biochemistry |
| David Harris, Ph.D. | Professor & Chairman | Biochemistry |
| Angela Ho, Ph.D. * | Assistant Professor | Biology |
| Tsuneya Ikezu, M.D., Ph.D.* | Professor | Pharmacology |
| Gyungah Jun, Ph.D.* | Assistant Professor | Medicine, Ophthalmology, Biostatistics |
| Konstantin Kondror, Ph.D. * | Professor | Biochemistry |
| Mark Logue, Ph.D. | Research Assistant Professor | Medicine |
| Kathryn Lunetta, Ph.D. | Professor | Biostatistics |
| Ann McKee, M.D. | Professor | Neurology |
| Peter Morin, M.D., Ph.D.* | Assistant Professor | Neurology, Biochemistry |
| George Murphy, Ph.D. * | Assistant Professor | Medicine |
| Wendy Qiu, M.D. | Associate Professor | Psychiatry, Pharmacology |
| Matthew Seaman, Ph.D. * | Senior Lecturer | University of Cambridge |
| Michael Sherman, Ph.D. | Professor | Biochemistry |
| Richard Sherva, Ph.D. | Research Assistant Professor | Medicine |
| Orian Shirihai, M.D., Ph.D. | Associate Professor | Medicine |
| Peter St. George-Hyslop, MD * | Professor | University of Toronto / Univ. Cambridge |
| Christopher Sullivan, Ph.D. | Instructor | Neurology |
| Ben Wolozin, M.D., Ph.D. * | Professor | Pharmacology |
| Vassilis Zannis, Ph.D. | Professor | Medicine, Biochemistry |
PROJECTS 2009-2010:
One thrust of the ARC will consider the genetic, genomic and cell biological aspects of the relationship of trafficking proteins to AD. Another project will assess APP metabolism and endosomal transport in primary hippocampal neurons from AppSwe/PS1ΔE9 mice using immunohistochemistry and confocal microscopy. A third project will investigate the role of Klotho (a protein which declines with age in rhesus monkey, rats and mice brains and and is decreased in mouse models of AD) on trafficking and its interaction with APP and SORL1. Other projects investigating other trafficking proteins in AD and trafficking in Parkinson disease will be developed.
In one project, Dr. Farrer and Dr. Baldwin will employ mega-base sequencing to follow-up association findings from analysis of many large whole genome wide association study datasets. Mega-base sequencing is more comprehensive (virtually all SNPs evaluated) and novel SNPs (ie, those not reported in public data bases will be identified). Specifically, we will sequence 2 genes (SORL1 and ACE which are firmly established as AD risk factors) in 5 AD cases and 5 control subjects from our study of AD in Wadi Ara, an Arab community in northern Israel. Since this is a genetic isolate/inbred community, we would expect lower genetic complexity making it easier to identify the causative variants. The sequence data will be analyzed using a variety of bioinformatics tools.
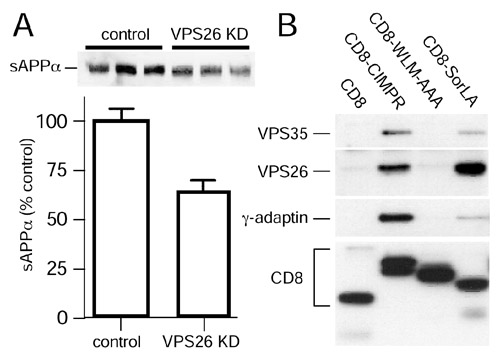
Previous data from Dr. Morin’s laboratory indicate that knockdown of Vps35, a retromer subunit containing cargo recognition information, results in defective retromer transport of APP and massive accumulation of cation- independent mannose-6-phosphate receptor in HEK-293 and SH-SY5Y cells over-expressing APPSwe. Using the same lentiviral shRNA-mediated knockdown of Vps35, in a second project Dr. Morin and Dr. Ho will examine through a series of steps the effects of retromer malfunction in a double transgenic APPSwe/PS1ΔE9 mouse line, which exhibit Aβ pathology as early as 4-6 months of age. Initial studies will assess APP metabolism and endosomal transport in primary hippocampal neurons cultured from APPSwe/PS1ΔE9 mice. Cells will be plated at high-density (100,000 cells/cm2) and Aâ secretion will be measured by ELISA. Cells plated at low density (10,000/cm2) will be analyzed by confocal microscopy to assess co-localization of APP and endo-lysosomal proteins (EEA1, Rab5, Tsg101, and LAMP1). Identification of abnormal APP metabolism in cultured neurons will further support studies in APPSwe/PS1ΔE9 mice. Lentiviral vectors expressing Vps35 shRNA or a GFP marker will be injected intraventricularly in newborn transgenic APPSwe/PS1ΔE9 and control mice. Cortical amyloid deposition, cellular APP and SorL1 distribution will be assessed by immunohistological methods and confocal microscopy, respectively, at 6 months of age.
The effect of the KL C370S and F352V variants on the trafficking of both secreted and transmembrane KL will be investigated in a third project by Dr. Abraham and Dr. Chen using western blotting, subcellular fractionation, immunocyto-chemistry, and Gaussia luciferase assays to detect secretion. By gaining insight into the trafficking of KL to the PM, they hope to find ways to increase the trafficking and shedding of KL, which is associated with increased lifespan. Since the intracellular trafficking of APP is affected by SORL1, they will determine whether KL is also a substrate for SORL1 and whether the human KL polymorphisms shown to affect its trafficking and secretion are differentially recognized and processed by SORL1. Another possibility exists that SORL1 affects the secretases that process KL. To test this hypothesis Drs. Abraham and Chen will (1) Examine the effects of Klotho polymorphisms on its trafficking and shedding; (2) Examine the effects of SORL1 overexpression or knock down on the various Klotho isoforms and the α secretases trafficking and shedding; (3) Determine the mechanism by which insulin activates secretion of Klotho and APP and involvement of SORL1 in this process; (4) Test whether Klotho modifes enzymatically (as a sialidase) APP, SORL1 or the α secretases; and (5) Test novel protein trafficking-related genes identified in the GWA studies by Dr. Farrer’s group.
Research Progress 2009-2010:
The ARC structure provided our group with both important resources and a broad collaborative network that integrates genetic, cell biological and animal model strategies for evaluating AD-related endosomal processes. There have been numerous and productive interactions among faculty with various expertise and perspectives. Examples of such interactions include the collaborations among ARC members on ARC-related projects (both funded and non-funded) are: (1) Carmela Abraham, Lindsay Farrer, Kathy Lunetta, Clint Baldwin, Matthew Seaman and Peter St George-Hyslop leading to a large R01 grant application; (2) Lindsay Farrer, Clint Baldwin, Carmela Abraham and George Murphy on neuronal iPS cells leading to a funded R21 grant; (3) Angela Ho (an AD researcher using transgenic mice) and Peter Morin (neurologist and biochemist working on retromer-related genes); and (4) Peter Morin and Ben Wolozin (a neurobiologist working on AD and Parkinson disease). Some of the most important discoveries and advances during the 2009-2010 funding period are as follows. We demonstrated that all of the vps10-domain containing (i.e., sortilin) proteins are genetically associated with AD and affect APP processing. Significant evidence of association was also obtained for several retromer-associated proteins including KIAA1033, Snx1, Snx3 and Rab7A. In parallel with these efforts, we showed that Snx3 and Rab7A interact with the cargo-selective retromer complex to regulate the membrane association of retromer and thereby are key mediators of retromer function. We also showed that SORL1 increases the secretion of Klotho. Since Klotho protects cells and organs from oxidative stress and cancer, and the entire organism from aging and cognitive decline, and since the vast majority of Klotho functions in its secreted form, any molecule that enhances secretion would be beneficial. The results indicated that we might be able to modulate Klotho secretion by regulating SORL1 expression. Finally, we found that Fzd5 significantly reduces Aβ-42 (the toxic form of amyloid that aggregates in AD brains) secreted from cells stably over-expressing APP.
Research Progress 2010-2011:
Lindsay Farrer and Peter St. George-Hyslop reported results from a series of genetic association and gene expression experiments showing that SORCS1 modulates APP processing and variants in this gene are associated with AD risk. Subsequently, they showed that several SNPs in each of SORCS2, SORCS3 and SORT1 were significantly associated with AD in Caucasians and other populations (0.001<p<0.05). They also demonstrated that expression of SORCS3 was lower in amygdala tissue from AD brains compared to control brains, but knockdown of all three genes using shRNAs in HEK293 cells caused a significant increase greater than 3-fold in detect γ-secretase processing of APP (p < 0.001 to p< 0.05) as compared to scrambled shRNA condition. In addition, they evaluated association of AD with 451 SNPs in 8 genes encoding the subunits of the mammalian retromer complex and 7 other retromer-associated genes emerging from Dr. Seaman’s studies in a sample of 8,309 AD cases and 7,366 controls. They obtained significant results at the SNP level or gene level with KIAA1033, SNX1, SNX3 and RAB7A. In parallel with these efforts, experiments conducted by Dr. Seaman showed that Snx3 and Rab7A proteins interact with the cargo-selective retromer complex to regulate the membrane association of retromer and thereby are key mediators of retromer function. Furthermore, although loss of Snx3 or Rab7A function both result in displacement of retromer from the endosomal membrane, we showed that these proteins operate by independent mechanisms.
Peter Morin and Angela Ho evaluated the effects of retromer disruption on AD in cultured primary neurons and mouse brain. Initial data indicated that knockdown of Vps35, a retromer subunit containing cargo recognition information, results in defective retromer transport of APP and massive accumulation of cation-independent mannose-6-phosphate receptor in HEK-293 and SH-SY5Y cells over-expressing APPSwe. They assessed Ab secretion (by ELISA) and APP localization using confocal microscopy in HEK-293 and neuroblastoma cells. Using the same lentiviral shRNA-mediated knockdown of Vps35, they began testing the effects of retromer malfunction in primary neurons cultured from APPSwe/PS1DE9 double transgenic mice. These mice exhibit amyloid pathology as early as 4-6 months of age. To assess cortical amyloid deposition in APPSwe/PS1DE9 mice, lentiviral vectors expressing shRNA or control RNA were injected intraventricularly in newborn transgenic animals to express Vps35 shRNA and a GFP marker. Following a suitable interval of 4-6 months, amyloid deposition was assessed neuropathologically.
Carmela Abraham and CiDi Chen conducted functional experiments on genes implicated in AD through genetic association studies in order to generate preliminary data for a large R01 application with Drs. Farrer, Baldwin and Seaman. They tested whether genes identified by Dr. Farrer’s group working independently or with the AD Genetics Consortium are involved in APP processing and Aβ production. They found that Sorl1 and Fzd5 significantly reduced Aβ42 secreted from cells stably over-expressing APP. They also confirmed the effect of Sorl1 on APP processing and Aβ42 secretion. In addition, they found that Fzd5 may affect Aβ42 secretion, which is a novel finding. Additional details of this work are described in the 2011 Annual Report.
Research Progress 2011-2012:
Previously, we demonstrated that most common SNPs associated with AD in Caucasians are not risk factors for the disorder in African Americans (AAs). To identify functional AD risk variants that are enriched in AAs, Drs. Farrer and Baldwin performed whole exome sequencing (WES) in 7 AA AD cases from the MIRAGE Study, which consists of primarily discordant sibling pairs. Variant calling with GATK revealed 431 novel (according to dbSNP build 131) non-synonymous SNPs that were observed in more than one subject. Of these, 63 SNPs from 58 genes were prioritized for genotyping based on predicted functionality, potential relevance to AD, and gene-network analysis. Forty-four of these variants were successfully genotyped in 435 AA cases and 422 cognitively normal age-matched AA controls from MIRAGE and another AA case-control cohort. Nominally significant associations were observed with several SNPs including two missense mutations (one predicted to be damaging and the other predicted to be benign) in AKAP9 (p=0.013 and 0.038 respectively). These SNPs have not been observed in white non-Hispanics and are present in ~ 1% of AAs according to the ESP database. All AKAP9 mutations were observed on the same background haplotype which occurs in 5% of AAs suggesting a common origin. Five other subjects (4 cases and 1 control) had the same background haplotype and only the pathogenic mutation. All instances of the mutations were verified by Sanger sequencing. AKAP9 encodes a kinase anchor protein which is expressed in the hippocampus, cerebellum, and the cerebral cortex.
Drs. Morin and Wolozin tested the hypothesize that variants in retromer proteins lead to abnormal trafficking of endocytosed protein aggregates/oligomers by examining the effects of retromer proteins on the uptake and trafficking of labeled oligomeric Ab. They identified shRNA sequences and lentiviral vectors. The vectors have been used to knockdown a test gene in rat primary neurons. These vectors were used to knockdown SorL1 and SorCS3. Knockdown is effective, but they have been unable thus far to achieve sufficient transduction efficiencies to perform experiments with fluorescently labeled Ab.
We hypothesized that specific polymorphisms in genes associated with AD and will affect the processing of APP by directing it to either the amyloidogenic or non-amyloidogenic pathway, thus affecting Aβ production. Dr. Abraham’s lab tested whether genes identified by Dr. Farrer’s group working independently or with the AD Genetics Consortium are involved in APP processing and Aβ production. To determine whether the PLXNA4 gene is involved in APP processing and Aβ production we transfected either empty vector or PLXNA4-Myc into cells stably expressing APP751 and analyzed APPsββ and Aβ 40 and 42 released into the medium. Over-expressing PLXNA4 did not affect APPsa secretion or Aβ42 and Aβ40 production (see Figure 1 in 2011-2012 report). These results suggest that PLXNA4 does not affect APP processing and/or Aβ production in our system.
Proper functioning of cells depends on the complex interplay between intracellular membrane trafficking pathways. Results from Dr. Kandror’s and other laboratories suggest that the sorting receptor sortilin plays the central role in endosome-TGN shuttling. The current dogma implicates different proteins in endosomal targeting and retrieval. He has put forward a novel hypothesis that the same multi-ligand protein receptor sortilin participates in both processes. In order to test this hypothesis, Dr. Kandror isolated intracellular transport vesicles that contain sortilin, and identified all component proteins in this compartment using silver staining and mass-spec analysis. To this end, his lab prepared a cell line that stably expresses sortilin tagged with myc and His epitopes at the C-terminus. They purified the total fraction of intracellular transport vesicles using continuous sucrose gradient centrifugation, and isolated sortilin-containing vesicles by immunoadsorption on Ni+-beads.
ARC AS A RESOURCE:
This ARC is a nidus for multi disciplinary research of neurodegenerative diseases, especially AD. Projects within the ARC will employ several high-throughput technologies including SNP-chip genotyping (Illumina platform), next-generation sequencing, and siRNA knockdown screening. Genetic and genomic data are managed and archived using state-of-the-art storage devices and capturing schema, and analyzed using BUMC’s high-performance computing system for genetics and genomics (LinGA). Members of the ARC include experts in molecular genetics, genetic epidemiology and statistical genetics, transgenic mouse models, neurology, neuropathology, neuroimaging, neurobiology, protein chemistry, proteomics, mitochondrial biology and genetics, bioinformatics, and systems biology.
Pictures, Images, Figures:
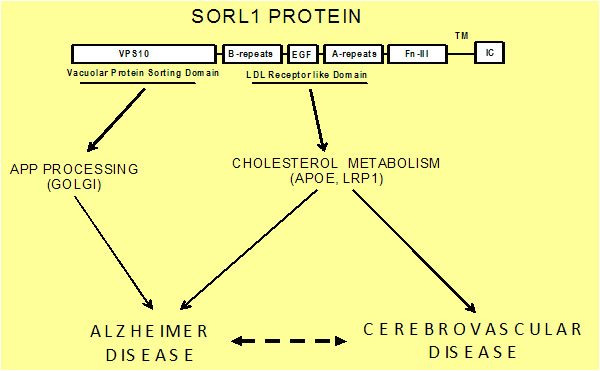
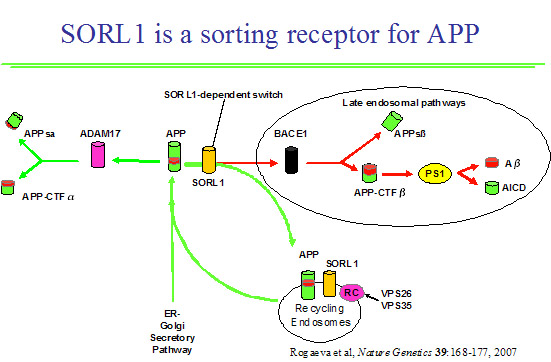
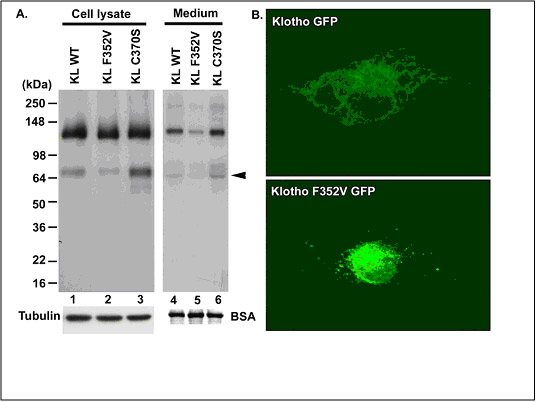
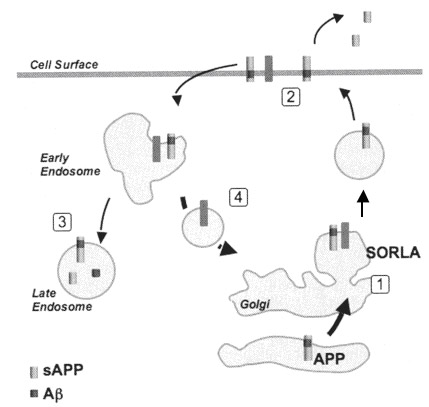
The role of Sorl1 in mediating APP localization and processing. Sorl1 associates with APP in the Golgi (1). APP and Sorl1 are endocytosed from the cell surface (2). In the absence of Sorl1, APP is delivered to a late-endosome where it can be cleaved to produce A peptides (3). At the early endosome, the retromer complex sorts Sorl1 (possibly with APP) into the endosome-to-Golgi retrieval pathway (4). Sorl1 can regulate APP localization by ‘trapping’ APP in the Golgi and/or by directing APP into the endosome-to-Golgi retrieval pathway. Image adapted from Willnow et al., 2009