Programmed Problem Set on Pharmacokinetics
Karen M. Harnett, Ph.D.
Assistant Professor of Pharmacology & Experimental Therapeutics
Boston University School of Medicine
Questions or comments should be mailed to Karen Harnett.
Return to Pharmacology Problem Sets
This programmed problem set is designed to help you learn and apply the concepts and vocabulary in Pharmacokinetics that have been introduced to you in lectures and text. The primary terms that you should master from this exercise are Half-life (t1/2), Volume of Distribution (Vd), Renal Clearance (ClR), Total Clearance (ClT), and Bioavailability (F).
Their definitions and methods for calculating these parameters are presented in the Pharmacology Glossary posted online and the lecture syllabus. You should complete this program before the first Pharmacology Discussion Session. Bring to that class any questions that arise during your use of this program. The first Discussion Session is designed to reinforce and expand upon the groundwork in pharmacokinetics presented in this problem set.
In order to help organize your interpretation of the data, a summary table is available for your use to enter in pharmacokinetic parameters as you determine them. Click here now to bring up the summary table page. Fill in the table as you work through the problem set, and then bring it with you to the discussion session on pharmacokinetics.
Chloramphenicol is a clinically useful antibiotic with the following chemical structure:
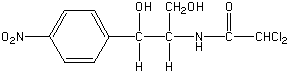
The compound is uncharged over the pH range 2-9 and is poorly soluble in water.
The concentration of chloramphenicol added to biological fluids may be determined in several ways:
- Bioassay: determination of the capacity of a sample to inhibit growth of microorganisms in vitro as compared to the capacity of a series of standards containing known amounts of chloramphenicol. Biotransformation products of chloramphenicol, produced in man and experimental animals, are inactive against microorganisms, so a bioassay of a sample containing both chloramphenicol and its metabolites measures unchanged chloramphenicol only. (Why would such an assay for chloramphenicol possibly be inappropriate in a patient receiving multiple antibiotic agents?)
- Chemical assay for nitro groups: NO2 groups are not present in mammalian fluids, so a chemical assay for this substituent can be used to quantitate chloramphenicol. But there is a problem with the accuracy, specifically the validity, of this assay because it detects all derivatives of chloramphenicol formed in vivo in which the NO2 remains but other parts of the molecule are modified. (Where might this molecule be modified? HINT: Might the aliphatic hydroxyl group be a site for glucuronide conjugation?)
- High-performance liquid chromatography (HPLC) or radioenzymatic assays: the advantage of these procedures is their capacity to assay specifically the concentration of the chloramphenicol molecule by chemical methods, and to distinguish chloramphenicol from its major metabolite (chloramphenicol glucuronide) and from inactive esteratic prodrug forms (chloramphenicol succinate or palmitate).
For this programmed problem set, you will be analyzing preclinical data collected during the initial development of this drug. The biological disposition of chloramphenicol was studied in dogs, as the animal model for preclinical pharmacokinetic studies. Experiments from two normal male dogs are presented below.
One animal, weighing 16.5 kg, received a single dose of 50 mg/kg (825 mg total) intravenously (i.v.); the second, weighing 18.0 kg, received 50 mg/kg (900 mg total) of chloramphenicol orally (p.o.). For both animals, serum and urine samples were collected at various times after drug administration and analyzed by bioassay for their content of microbiologically active material (unchanged chloramphenicol). (These data are taken from the studies published in the Journal of Pharmacology and Experimental Therapeutics by the investigators at Parke-Davis who were responsible for the preclinical development of this drug prior to its marketing.)
| Time after administration of chloramphenicol (hr): |
0.5 |
1.0 |
1.5 |
2.0 |
3.0 |
4.0 |
6.0 |
8.0 |
| Serum concentration of chloramphenicol (µg/ml): |
|
After i.v. administration |
32.2 |
24.2 |
17.0 |
12.5 |
6.6 |
3.5 |
0 |
–* |
|
After p.o. administration |
0 |
1.0 |
— |
10.0 |
— |
15.0 |
8.7 |
4.5 |
* indicates not determined
Complete urine collections were made during the period following administration of chloramphenicol. The concentration of chloramphenicol in each urine sample was determined and multiplied by urine volume to determine the amount of drug excreted in the urine in each interval (see below).
| Time after administration of chloramphenicol (hr): |
0-2 |
2-4 |
4-6 |
6-8 |
8-24 |
0-24 total amt
(% of dose) |
| Amount of chloramphenicol in urine (mg): |
|
After i.v. administration |
55 |
17 |
5 |
— |
— |
77
(9.3%) |
|
After p.o. administration |
6 |
24 |
12 |
9 |
16 |
67
(7.4%) |
The following program will guide you through an evaluation of the data to conclusions about the pharmacokinetics of chloramphenicol. Your first step for each question should be to inspect the data and estimate the pharmacokinetic parameter required to arrive at the answer. These are the skills you will most often use in interpreting clinical data or the clinical literature. You may better understand these data if you graph them and complete a regression analysis of ln Cp vs. time. Use the Cp vs Time spreadsheet and enter the plasma concentration (Cp) data from above, before moving on to Item I. If you find that you have difficulty working with these data, please consult a faculty member or tutor before the first discussion session.
NOW GO TO ITEM I
I. The elimination of chloramphenicol from the serum after it is given i.v. conforms to the laws of:
a. Zero-order kinetics
No; a drug is eliminated according to zero-order kinetics if a constant amount is eliminated in equivalent units of time, regardless of the amount of drug present to be eliminated. In this case, the amount eliminated from the serum decreases in each successive 30-minute period. Please go back to Item I.
b. First-order kinetics
Yes; inspection of the serum concentrations (Cp) of chloramphenicol indicated that in successive 30-minute periods after i.v. administration the percent decrease is constant, whereas the absolute decrease becomes smaller. Your graph of the data with ln of Cp on the ordinate (y-axis) and time on the abscissa (x-axis) is linear. (If you had not made the logarithmic transformation of Cp and had graphed Cp vs. t, your data would be clearly curvilinear.) These observations are consistent with elimination by first-order kinetics.
Note that, according to the definition of first-order kinetics, the rate of elimination of drug from plasma is proportional to the plasma concentration. This statement is describable as: ΔCp / Δt = kelCp. The integral of this differential equation is lnCp = lnC0 – kelt, where C0 is the plasma concentration at zero time. So data conforming to first-order elimination are described by an equation of this form. As in this case, with chloramphenicol, a plot of ln Cp vs. t is a straight line with an intercept on the ordinate of ln C0 and a slope of kel, the elimination rate constant. . Note that the elimination t1/2 = 0.693/kel Now proceed to Item II.
c. Mixed-order kinetics (a combination of zero-order and first-order kinetics)
No; perhaps you should plot the serum data? What does the shape of the curve tell you about the kinetics of elimination? How did you rectify the curve, i.e., transform the data in order to generate a straight line? Go back to Item I.
II. The elimination half-life of chloramphenicol is:
a. About 30 minutes
No; the subject’s serum was sampled and assayed for chloramphenicol at 30 minute intervals initially, but this is not the t1/2 value. Do you understand how to determine the t1/2? Determine the time interval for the concentration to drop 50%. Try Item II again.
b. About 1 hour
Yes; since the elimination of chloramphenicol conforms to the laws of first-order kinetics, the data are described by the following equation:
ln Cp = ln C0 – kel
where kel, the slope of the plot of ln plasma concentration vs. time, is the fraction of the drug eliminated per unit time. The half-life is the time it takes to decrease the concentration (C) by half (1/2C).
Consequently, the half-life t1/2 = [ln (C/ 1/2C)]/kel = [ln 2]/kel = 0.693/kel. Thus:
t1/2 = 0.693/kel
Since kel = (ln C1 – ln C2)/(t1 – t2) = 0.64/hr, then t1/2 = 1.1 hr. By inspecting the data, you can see that Cp decreases to about half its value in 1 hour. Now go on to Item III.
c. Neither of the above
No; Inspect the plasma concentrations after i.v. administration. How long does it take for the levels to drop by one half? Choose any serum concentration of X. Find the time t1 at which X occurs. Then find the time t2 corresponding to 1/2X. The serum half-life is t2 – t1, the time required for reduction of the serum concentration by one-half. [If you made a plot of ln serum concentration vs. time, check your calculations of the slope kel. Then reconsider the relationship between kel and t1/2 (t1/2 = 0.693 / kel). Try Item II again.
III. In the 16.5 kg dog the apparent volume of distribution (Vd) of chloramphenicol is:
a. Less than 1 liter
No; reconsider the total dose given to this 16.5 kg subject. Go back to Item III.
b. About 4 liters
No; think again about the total dose given to this subject and then about the appropriate equation for determining the volume of distribution. Go back to Item III.
c. About 18 liters
Correct, you have extrapolated the serum concentration to zero time after i.v. administration, 45 µg/ml (anti-ln of 3.8), and divided this number into the total dose, 825 mg, to determine Vd in liters, 18.3 l. Or you estimated the zero time level at about 48 µg/ml by realizing that the level is 24µg/ml at 1 hr after administration and because the half-life is about 1 hr, the zero time level would be two times higher. Now proceed to Item IV.
d. About 50 liters
No; you may have arrived at an erroneous value by attempting to establish a zero-time serum concentration from the data following oral administration. The extrapolation to zero-time can be valid only with serum data having no substantial absorption phase; consider the data following i.v. administration. Go back to Item III.
e. None of the above
No; The extrapolated zero-time value on your graph of ln Cp vs. time should be about 3.80. The anti-ln of this number is about 45 µg/ml. Or estimate the zero-time value as about 48 µg/ml, given a half-life of about 1 hr and a Cp value of about 24µg/ml at 1 hr. Now try again to calculate Vd. Go back to Item III.
IV. In the dog, as in a human adult male of average body weight and composition, the volumes of plasma, extracellular and total body water average 4%, 17%, and 58% of body weight, or 0.04, 0.17, and 0.58 l/kg respectively. Therefore, the apparent volume of distribution of chloramphenicol in the dog corresponds approximately to which of the following body fluid compartments:
a. Plasma volume
No; divide the Vd of chloramphenicol (18.3 l, see comment to Item IIIc) by the weight of the subject (16.5 kg) and compare the Vd in l/kg to that of plasma (0.04 l/kg or 4% of body weight). Go back to Item IV.
b. Intracellular fluid volume
No; would you anticipate that a drug which is administered into the vasculature and which passes across vascular walls would not be distributed also to the extracellular fluid? Non-uniform distribution of the drug might result in a calculated volume corresponding to the intracellular fluid space – but that’s not true in this case! Go back to Item IV.
c. Extracellular fluid volume
No; A Vd of 18.3 l in a 16.5 kg subject is substantially greater than 17% of body weight, which is the approximate size of the extracellular fluid volume. Go back to Item IV.
d. Total body water
No; Total body water is about 58% of body weight which in this subject would be 9.6 liters. Go back to Item IV.
e. Greater than total body water
Correct; 18.3 l represents 1.1 l/kg body weight or 110% of the total body weight of this subject, considerably higher than the average value of about 58% for total fluid volume. The drug may not distribute uniformly; sequestering at some site outside the plasma volume would reduce the value of the initial serum concentration and increase the apparent volume of distribution. Because the calculated Vd is so high, it is reasonable to conclude that choramphenicol readily crosses membranes and distributes into cells and fluid spaces such as the CSF. Now proceed to Item V.
V. Analysis of the serum concentration data for chloramphenicol after its i.v. and oral administration suggests that:
a. The elimination half-life of chloramphenicol depends on the route of administration
No; the elimination half-life is determined by the total clearance (ClT) and the volume of distribution, t1/2el = 0.693 Vd/ClT.
ClT and Vd generally are the same for different routes of administration (please make sure that makes sense to you). So the elimination half-life is usually the same for different routes of administration.
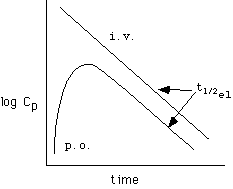
If the absorption half-life (t1/2abs) is shorter than the elimination half-life (t1/2el) , then the terminal portion of the log Cp vs. t curve after p.o. administration will parallel the curve after i.v. administration and in both cases are indicative of the elimination half-life, as shown in the top curve:
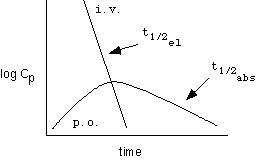
But in cases where the elimination is very rapid, as with chloramphenicol, the terminal curves may not be parallel because the kinetics of decline after p.o. administration are indicative of the absorption half-life, not the elimination half-life, as shown below:
Please return to Item V to find a correct answer.
b. The absorption of chloramphenicol into the systemic circulation is incomplete, since peak concentration is lower after p.o. than i.v. administration
No; your logic is not quite right. After administration of a drug by any route other than i.v., the peak concentration is determined by the absorption half-life, elimination half-life, the extent of absorption (bioavailability), the dose, and the volume of distribution. Generally, the elimination half-life and Vd are about the same regardless of the route of administration. But the slower the rate of absorption, the lower the peak concentration even when the entire dose is absorbed, i.e., when the bioavailability is 100%. Try Item V again.
c. The absorption of chloramphenicol is complete, since the area under the concentration vs. time curve after the p.o. route (83 mg hr/ml) is about equivalent to that after i.v. administration (70 mg hr/ml)*
Correct: To quantitate the percent of the dose absorbed into the systemic circulation, the bioavailability (F), you can measure the area under the curve of serum concentration vs. time (AUC) for the p.o. route and compare this value to the area generated after i.v. administration. This approach is the standard procedure for assessing the bioavailability of formulations of a drug for oral use that may differ with respect to factors such as disintegration time, dissolution rate, stability, etc.
In this case, the AUCp.o. is 83 mg.hr/ml and the AUCi.v. is 70 mg.hr/ml, so the bioavailability appears to be 100%. Strictly speaking, 83/70 x 100% = 116% bioavailability. Theoretically, the bioavailability cannot exceed 100%. But this study was carried out in two dogs, each of which received the drug by only one route. AUC is determined by dose, bioavailability, and total clearance. Total clearance may have been slightly lower in the dog receiving the p.o. dose, in which case the estimate of bioavailability would be a little high. The preferred experimental design for studies of bioavailability entails administration of the same total dose of drug by both routes in each subject (cross-over design) with sufficient time between the doses to avoid possible drug accumulation or drug-induced changes in its own pharmacokinetics. This cross-over design minimizes differences in total clearance.
Please look for another correct answer in Item V. If you have already found it, proceed to Item VI.
d. The absorption of chloramphenicol after oral administration exhibited a lag phase and was not rapid in onset
Yes; measurable serum levels were not detectable until 1 hour after the oral dose, and the peak concentration did not occur until 4 hours. The oral dose was administered in gelatin capsules. Possibly, capsule disintegration, dissolution of chloramphenicol powder, and emptying of the drug from the stomach into the small intestine affected the rate of chloramphenicol absorption. Since chloramphenicol is an uncharged molecule that appears to readily distribute into cells, it is unlikely that the rate-limiting factor in its absorption is passage of dissolved drug through the intestinal mucosa. Please look for another correct answer in Item V. If you have already found it and the comment to Item Va, which has a helpful figure, then time for a new challenge! Now try Item VI.
(*Note that you need not verify these area values; they have been correctly estimated.)
VI. The excretion of chloramphenicol in the urine following i.v. as compared to oral administration suggests that:
a. Regardless of the route of administration, chloramphenicol is primarily eliminated from the body by a mechanism other than renal excretion
Yes; since only 7.4% and 9.3% are recovered in the urine after p.o. and i.v. administration respectively, most of the dose of chloramphenicol must be eliminated by another mechanism. Return to Item VI to see if there is another correct answer.
b. Chloramphenicol is incompletely absorbed from the intestine
No; The total recovery of chloramphenicol in the urine, 0-24 hr after oral administration, is low, 7.4% of the dose. But this value is very similar to the cumulative urinary recovery after i.v. administration, 9.3% of the dose, indicative of nearly complete bioavailability. Reconsider the choices in Item VI.
c. Chloramphenicol is incompletely absorbed and/or is eliminated by first-pass biotransformation in the liver
No; the cumulative excretion of unchanged chloramphenicol after p.o. administration (67 mg/900 mg = 7.4% of dose) is similar to that after the i.v. route (77 mg/825 mg = 9.3% of dose). The bioavailability of chloramphenicol appears to be high. Your conclusion, based on cumulative urinary excretion data, agrees with your previous one based on AUCs of the serum data (see the comment to Item Vc), as you would expect. Since the renal excretion of chloramphenicol is so low, it would be preferable to have data on the urinary excretion of both unchanged drug AND any metabolites after p.o. and i.v. administration. Then you could estimate the fraction of the dose which is absorbed by dividing the cumulative excretion of drug AND metabolites after p.o. administration by the corresponding value after i.v. administration. Try Item VI again.
d. Urine collected 6-24 hr. after i.v. administration of chloramphenicol would be expected to contain substantial amounts of unchanged drug
No; generally the excretion rate of unchanged drug into urine is proportional to the plasma or serum concentration. You can see from the data that by 6 hours after i.v. administration the concentration of chloramphenicol in serum dropped to below measurable levels. Remember also that the elimination half-life is 1.1 hr in this animal. One half-life after an i.v. dose only 50% is remaining in the body, two half-lives only 25%, three half-lives only 12.5%, etc., so by 6 hours less than 3% is left. Of that 3% only a minor fraction is eliminated by the kidney, so you wouldn’t expect to recover much in the urine after 6 hours. If you have considered choices 1-4 and found only one correct answer, you may now proceed to Item VII.
VII. Some understanding of the mechanisms by which a drug is cleared from the plasma into the urine can be acquired by calculating of the renal clearance. The renal clearance (ClR) indicates the volume of the plasma that must have been cleared of the drug in order to achieve the output observed in the urine. Recall your calculations of renal clearance of nutrients such as glucose in physiology.
| ClR (ml/min) |
= |
rate of renal excretion (amount/min) |
| plasma concentration (amount/ml) |
(Note that usually the concentrations of a drug in serum and plasma are about the same.)
| ClR |
70-kg human |
16.5-kg dog |
Estimate for: |
| Inulin |
120 ml/min |
50 ml/min |
GFR |
| Para-aminohippuric acid |
650 ml/min |
200 ml/min |
RPF |
The renal clearance of chloramphenicol in the dog after i.v. administration is:
a. 0 to 9 ml / min
No; Have you chosen the appropriate serum concentration for your calculation, the value on the graph at the midpoint of the urine collection period, e.g., urine output from 0 to 2 hour, serum concentration at 1.0 hr? Redo your calculation for Item VII.
b. 10 to 35 ml / min
Correct;
Based on 0-2 hr urinary recovery:
| ClR |
= |
Excretion rate of drug=55 mg / 120 min |
| Cp at 1 hr |
24.2 µg/ml |
|
|
= |
(0.019 mg•ml) |
•(1000 µg) |
|
| µg•min |
mg |
= 19 ml/min |
Based on 2-4 hr urinary recovery:
| ClR |
= |
17 mg / 120 min |
= 21.5 ml/min |
| 6.6 µg/ml |
Note that your two estimates of ClR of chloramphenicol are very similar, as you would expect. Generally, the renal clearance is constant and not dependent on the dose of the drug or the time after drug administration. However, often experimental estimates of ClR may be quite variable. One cause of variability is incomplete collection of urine excreted during the study interval. Can you think of other sources of variability? Proceed to Item VIII.
c. 36 to 54 ml / min
No; Try Item VII again. Carefully follow the units to the values you are using in this calculation.
d. 55 to 200 ml / min
No; have you chosen the appropriate serum concentration for your calculation? Have you divided the amount excreted by the minutes of collection? Go back to Item VII.
e. Greater than 200 ml / min
No; your calculation is incorrect – so is your understanding of the concept of renal clearance! This value cannot exceed renal plasma flow which is 200 ml/min in this subject. Try again and watch your labels carefully to help correctly compute ClR in ml/min. Go back to Item VII.
f. Likely to be significantly different than after oral administration
No; renal clearance indicates the volume of plasma, serum or blood per unit time that is completely cleared of drug by urinary excretion. The route by which the drug enters the circulatory system is generally irrelevant to the mechanism by which the kidney removes drug. You could compute ClR of this drug after p.o. administration to check this. Better yet, try to find the correct answer for Item VII.
VIII. The value of renal clearance of chloramphenicol in the dog indicated that:
a. Chloramphenicol is filtered by the glomeruli
Even without calculating the renal clearance, you can be fairly confident that this drug, and most drugs except protein therapeutics, are filtered by the glomeruli, if present in the plasma (and not 100% bound to plasma proteins!). However, this conclusion is incomplete; go back to Item VIII and consider another answer.
b. Chloramphenicol is incompletely filtered by the glomeruli and/or partially reabsorbed
Correct: In the dog the renal clearance of chloramphenicol, about 20 ml/min, is less than half that for inulin, about 50 ml/min. Two explanations for a renal clearance less than the glomerular filtration rate are: 1) Not all the drug is filtered out of the blood because of binding to plasma proteins. Chloramphenicol is reported to be about 50% bound at therapeutic concentrations. 2) Some of the filtered drug is reabsorbed from the tubules. You have found that chloramphenicol appears to pass readily through cell membranes, so it is reasonable that some of the drug would be reabsorbed. A definitive study to determine the mechanism of renal clearance of chloramphenicol would entail measurement of free chloramphenicol in serum and calculation of renal clearance based on these data. Now try Item IX.
c. Chloramphenicol is filtered, partially reabsorbed, and secreted
No; it is not possible to determine from a renal clearance value less than that of inulin whether tubular secretion has taken place. You can conclude that there is no NET tubular secretion. Go back to Item VIII.
IX. The rate of renal excretion of chloramphenicol and its renal clearance most likely:
a. Increase if the pH of the urine is decreased, as occurs with basic drugs such as amphetamine (pKa=10)
Recall that chloramphenicol is a neutral compound that does not become charged at physiologic pH. It is therefore unlike amphetamine, a primary amine that at lower pH takes on more hydrogen ions and is less well reabsorbed from the renal tubules. Try Item IX again.
b. Are unrelated to the pH of the urine
Yes; recovery of chloramphenicol in the urine (9% of the dose after i.v. administration) indicates that only a small amount of the drug is cleared from the blood by the kidney. The renal clearance indicates chloramphenicol may undergo partial tubular reabsorption. But the drug is a neutral compound that does not become ionized at physiologic pH. So the pH of the urine should not influence the degree of tubular reabsorption. Therefore the rate of renal excretion and renal clearance of chloramphenicol is NOT likely to be affected by renal pH. Proceed to Item X.
c. Increase if the pH of the urine increases, as occurs with acidic drugs such as salicyclic acid (pKa=3)
No; a drug in unionized form is more lipid soluble than its ionized counterpart. Reabsorption of unionized drug occurs more readily from the tubular fluid back into the blood. The extent of reabsorption is therefore dependent on the degree of ionization of the drug in the tubular fluid. Since salicylic acid has a pKa of 3, as the pH of the urine increases less of the drug is in unionized form. Consequently, reabsorption occurs more slowly and more of the drug is recovered in the urine. But chloramphenicol does not have a carboxylic acid group like salicylate and does not behave like an acid. Return to Item IX.
X. The total clearance (ClT) of a drug indicates the clearance from the body by all routes and mechanisms. This value can be estimated from:
ClT = (0.693/t1/2)Vd
(Suggestion: Use half-life in min and Vd in ml so your estimate of ClT is in ml/min. Do you get a value for chloramphenicol in the dog of about 195 ml/min?).
Or if kel has already been calculated, it can be computed from:
ClT = kel Vd, where kel is the slope of the ln Cp vs. t plot after i.v. administration.
Comparison of the total clearance of chloramphenicol (195ml/min) to its renal clearance (See comment to Item VIIIb) indicates that:
a. Chloramphenicol is cleared only by the kidney, and not by the liver or other organs
No; the total clearance of chloramphenicol, 195 ml/min, substantially exceeds the renal clearance, 20 ml/min, so additional routes of elimination must occur. Try Item X again.
b. Chloramphenicol is cleared by the liver, as well as by the kidney
Since the total clearance exceeds the renal clearance, you are correct in assuming clearance of chloramphenicol occurs by another organ. The liver is a good bet because of its capacity for biotransformation and biliary excretion. However, since you don’t have any data yet on chloramphenicol in bile or on metabolite formation by the liver, there is a better answer to Item X.
c. Chloramphenicol is cleared from the body partly by biotransformation
No; clearance of chloramphenicol by biotransformation may explain why the total clearance greatly exceeds the renal clearance. And you might expect that the hydroxyl groups are sites for glucuronide conjugation, mediated by glucuronyl transferases in the liver. However, since you don’t have any data on metabolites in the plasma, urine, or bile, this conclusion is not yet justified. For example, the nonrenal clearance could be attributable exclusively to biliary excretion of chloramphenicol without any metabolite formation. So there is still a better answer to Item X, and here’s your chance to find it! Go back to Item X.
d. Nonrenal mechanisms account for about 90% of the clearance of chloramphenicol
Yes; that is true. The total clearance, based on your estimates of half-life or kel (see comment to Item IIb) and Vd (see comment to Item IIc), is 195 ml/min; renal clearance (see comment to Item VIIb) is 20 ml/min. The difference of 175 ml/min, which is 90% of the total, must reflect clearance by nonrenal mechanisms. Note: As would be expected, this estimate of nonrenal clearance (as a percent of total clearance) agrees with the percent of the dose which is not recovered in the urine, 100% – 9% = 91% (see comment to Item VIa), and which must be eliminated by biotransformation and/or nonrenal excretion. Having mastered this item, now try Item XI.
XI. In this experiment the chemical assay for nitro groups was also used to determine serum and urine content of nitro-containing compounds (both chloramphenicol and its metabolites). For a given sample, total nitro content minus chloramphenicol content indicates metabolite content. The data for chloramphenicol glucuronide, the major metabolite, indicate a renal clearance of 96.3 ml/min and a cumulative urinary excretion of 60.5% of the chloramphenicol dose after i.v. administration in the 16.5 kg dog. These data suggest that:
a. Chloramphenicol glucuronide is eliminated in part by renal tubular secretion, but a considerable portion of the dose is eliminated by another mechanism, possibly in the bile
Yes; The ClR of the metabolite is greater than that of inulin in the dog, consistent with renal tubular secretion. This is not surprising since glucuronides are organic acids secreted by the carrier mechanisms in the proximal tubules. Glucuronide conjugates are also often secreted via the bile. Proceed to Item XII — the end is in sight!
b. The bacteriostatic effect of chloramphenicol in the urine is primarily caused by its metabolites
No; remember that chloramphenicol in this study was measured by a microbiological assay. Its metabolites, the primary one being the glucuronide conjugate, are inactive against microorganisms. Some drug metabolites are active, but glucuronides generally are not. The assay for total nitro compounds in urine may detect considerably more material than the bioassay because the inactive metabolites are extensively excreted by this route. Return to Item XI.
c. Renal impairment in a patient is more likely to increase the serum concentrations of chloramphenicol than of its metabolite
No; chloramphenicol appears to be extensively eliminated by biotransformation, and its metabolite is extensively cleared by renal tubular secretion into the urine. Go back to Item XI.
XII. In another experiment, the i.v. injection of 25 mg/kg of chloramphenicol to a 16.5 kg dog was found to produce bacteriostatic serum concentrations. In comparison to a dose of 50 mg/kg, this dose would probably result in:
a. A shorter elimination half-life
No; from the serum data provided you know that the elimination conforms to laws of first-order kinetics for amounts in the body that would be achieved by a 25 mg/kg dose. Consequently, the half-life is independent of the dose. You don’t know, however, whether this is true for doses greater than 50 mg/kg i.v. The higher the dose the greater the likelihood of saturating a mechanism of elimination mediated by enzymes. Go back to XII and make another choice.
b. A smaller renal clearance
No; you have no basis for making this conclusion. Furthermore, renal clearance is usually independent of dose. Make another choice.
c. A smaller volume of distribution
No; in the usual case the Vd of a drug does not change with dose. Go back to Item XII.
d. A halving of the duration of bacteriostatic effect in serum
No; for drugs administered as an iv bolus and eliminated by first-order processes, with a close correspondence between plasma concentrations and a reversible effect, the duration of action is not proportional to the dose, but is proportional to the log of the dose. It takes geometric increases in bolus doses to produce arithmetic increases in duration of action.
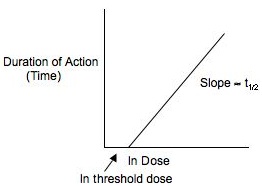
The time that elapses for the 50µg/kg amount in the body to fall to the 25 µg/kg level is equal to one elimination half-life. The duration of action is therefore one half-life less with the 25 µg/kg dose as compared to the 50 µg/kg dose. Similarly, you would predict that 100 µg/kg would produce a duration of action one half-life longer than the 50 µg/kg dose. Having pondered this a bit, you are ready to reconsider the correct answer to Item XII.
e. A 1-hr reduction of the duration of bacteriostatic effect in the serum
Yes; the duration of bacteriostatic effect is reduced by one elimination half-life, which for chloramphenicol is about 1 hour. If you haven’t already, you may find it useful to read the explanation for choice XIId. If you’ve considered this explanation, then charge forward to the LAST COMMENT below!
LAST COMMENT
You’ve now completed this program. Congratulations!
Come to the afternoon discussion session on pharmacokinetics, and we will use your skills in interpreting clinical pharmacokinetic data to consider the impact of disease, age and drug formulation and to design multiple dosing regimens.