A new paper in eLife from the Harris lab has uncovered a novel function for different domains of the prion protein. Bei Wu, an Instructor in the laboratory was lead author.
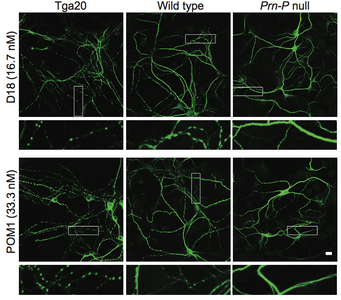
Prion diseases, or transmissible spongiform encephalopathies, comprise a group of fatal neurodegenerative disorders in humans and animals for which there are no effective treatments or cures. These diseases are caused by refolding of the cellular prion protein (PrPC) into an infectious isoform (PrPSc) that catalytically templates its abnormal conformation onto additional molecules of PrPC.A similar, prion-like process may play a role in other neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases and tauopathies, which are due to protein misfolding and aggregation. Here, using a combination of electrophysiological, cellular, and biophysical techniques, we show that the flexible, N-terminal domain of PrPC functions as a powerful toxicity-transducing effector whose activity is tightly regulated in cis by the globular C-terminal domain. Ligands binding to the N-terminal domain abolish the spontaneous ionic currents associated with neurotoxic mutants of PrP, and the isolated N-terminal domain induces currents when expressed in the absence of the C-terminal domain. Anti-PrP antibodies targeting epitopes in the C-terminal domain induce currents, and cause degeneration of dendrites on murine hippocampal neurons, effects that entirely dependent on the effector function of the N-terminus. NMR experiments demonstrate intramolecular docking between N- and C-terminal domains of PrPC, revealing a novel auto-inhibitory mechanism that regulates the functional activity of PrPC.