Alan Fine, M.D.
Faculty and Fellows
afine@bu.edu
Professor of Medicine
Director Responsible Conduct of Research Program (RCR) Boston University
Medical School: University of Michigan
Internship: Boston City Hospital
Residency: Boston City Hospital
Fellowship: Boston University, Harvard University
CLINICAL
General Pulmonary Medicine. Dr. Fine is a Professor in the Department of Medicine and attends on the general pulmonary consult service, medical intensive care unit and acute pulmonary care clinics at the Boston VA Medical Center.
RESEARCH INTERESTS
-
- Ciliated Cell Functions and Origins
- Lymphatic Development
- Bronchial Smooth Muscle Phenotype Regulation
- Research Ethics
Introduction
My laboratory has had long-standing NIH support and broadly speaking, been focused on elucidating the basic mechanisms that regulate lung repair and normal lung growth and development. My current projects are focused on: 1) the regulatory properties of retinoic acid on bronchial smooth muscle cell (BSM) phenotype 2) the mechanisms underlying lymphatic formation in the developing lung, and 3) the origin, phenotype, and functional properties of MIWI2 positive multi-ciliated cells. For all of these projects, we have developed unique mouse tools that allow for establishing lineage relationships and the isolation of select cell populations for genomic and bio-physiological studies.
1) Retinoic acid and BSM phenotype: In collaboration with Dr. Felicia Chen, we demonstrated that diet-derived retinoic acid plays a crucial role in maintaining bronchial smooth muscle (BSM) homeostasis in the adult lung. In several distinct live mouse and primary human cell systems, we found that active retinoic acid signaling is required to maintain BSM in a non-hyper-contractile and non-fibrotic state. We went on to demonstrate that this occurs through suppression of local TGF-β signaling. Without ongoing retinoic acid signaling, mice spontaneously develop an asthma-like syndrome characterized by hyper-responsiveness to broncho-constrictors; this condition is non-inflammatory, involves increased deposition of airway collagen, increased smooth muscle mass (Figure 1), and is sustained even after replenishment of retinoic acid. Future work is geared toward a further understanding of how specifically retinoic acid inhibits TGF-β pathway activation in normal BSM during homeostatic conditions, and what are the mechanisms involved in the long-term effect of transient retinoic acid insufficiency on BSM phenotype.
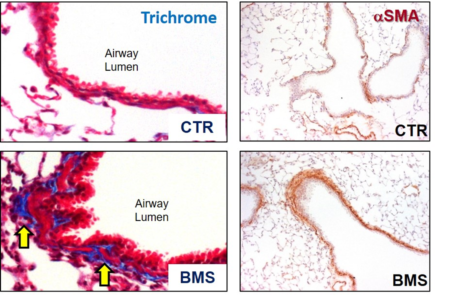
Figure 1: Airway Remodeling in mice fed a diet containing the pan-retinoic receptor inhibitor BMS for 5 days. Control mice (CTR) were fed a regular diet. Lung sections were trichrome-stained to evaluate for connective tissue (yellow arrows) or with an antibody to smooth muscle actin (αSMA) to assess overall BSM mass. Note increases in connective tissue and BSM in mice fed BMS.
2) Lymphatic Formation in the lung: A graduate student in my laboratory (Tim Norman) has been investigating the gene programs that regulate lung lymphatic endothelial cell (LEC) maturation just prior to birth. The rationale for this work is that the capacity for fluid removal and immune cell trafficking from the lung to lymph nodes are fundamental to the transition to air-breathing. The intent of these studies is to identify pathways that mediate acquisition of these key functions in LECs during the peri-natal period. Another facet of this work is focused on understanding how the lung’s lymphatic system grows and expands during early post-natal lung life. Using lineage tracing, our preliminary data suggest extensive remodeling of lymphatics with near complete replacement of fetal LECs during post-natal life from a colony stimulating factor receptor 1+ (CSF1R) progenitor (Figure 2). While CSF1R expression denotes cells of myeloid lineage, we are carefully evaluating for non-myeloid CSF1R+ cells, including rare LECS that might also serve as progenitors. In this context, we employed very sensitive in situ hybridization methodology and did not observe expression of endogenous CSF1R or Cre recombinase mRNAs in LECS during early post-natal life. Using lineage tracing, our preliminary data suggest extensive remodeling of lymphatics with near complete replacement of fetal LECs during post-natal life from a colony stimulating factor receptor 1+ (CSF1R) progenitor (Figure 2). While CSF1R expression denotes cells of myeloid lineage, we are carefully evaluating for non-myeloid CSF1R+ cells, including rare LECS that might also serve as progenitors. In this context, we employed very sensitive in situ hybridization methodology and did not observe expression of endogenous CSF1R or Cre recombinase mRNAs in LECS during early post-natal life. While we are performing additional studies, these findings, to date, are consistent with the presence of an exogenous CSF1R+ progenitor that replaces established fetal LECS during early post-natal lung development. How this may occur, the identity of the CSF1R+ progenitor, and the underlying differences between fetal and adult LECs are areas that we will explore in these novel studies.
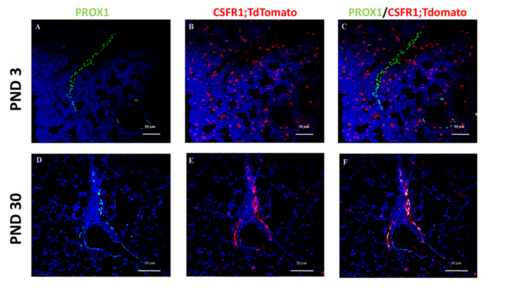
Figure 2: Derivation of lymphatic endothelial cells (LECs) from a CSF1R+ progenitor during post-natal lung development. Mice expressing a tamoxifen inducible Cre under the control of the colony stimulating factor 1 receptor 1 (CSF1R) promoter were crossed to mice that express a stop- floxed red fluorescent tdTomato gene. Progeny from this breeding were injected at post-natal days (PND) 1 and 2 before sacrifice at PND 3 or PND 30. Lung sections were stained with an antibody to Prox1 to identify LECs and with an antibody to tdTomato to identify cells that are derived from a CSF1R+ lineage. Note: tdTomato labeling of round macrophage cells but not LECs at PND 3. In contrast, there is dual labeling of LECs at PND 30, indicating derivation of cells from a CSF1R+ progenitor.
3) MIWI2 positive ciliated cells. In collaboration with Dr. Matt Jones, we examined the lung epithelial transcriptional responses to acute inflammation. In this work, we observed a significant increase in the mRNA expression of the Piwi protein, MIWI2. This protein had previously only been identified in germ cells where it functions to suppress transposon transcription. Localization studies showed that MIWI2 is selectively expressed in muliticilated cells in the normal adult mouse lung airway. We were surprised to find that MIWI2 expression was restricted to a subset (approximately 30%) of mouse ciliated cells (MPACs), a pattern recapitulated in the human lung. We also found that the number of MPACs increases during acute inflammation. Our preliminary ontogeny data provide support to the hypothesis that MIWI2-negative and MIWI2-positive ciliated cells are separate airway cell lineages. Transcriptomic and gene ontology analyses demonstrated that MPACs exhibit a distinct gene expression phenotype compared to ciliated cells not expressing Miwi2, further underscoring the novel concept of ciliated cell heterogeneity (Ref. 1, and Figure 3). Collectively, these findings suggest new hierarchical relationships in the airway epithelium. To evaluate for this possibility, we plan to elucidate the ontogeny, half-life, and origins of MPACs relative to non-MPACs. One important issue that we hope to clarify is the relative lineage relationship between MPACs and non-MPACs, themselves. Determining the precise roles of ciliated cell MIWI2 in regulating inflammatory states in the lung along with distinguishing the molecular and functional differences between MPACs and non-MPACs in inflammation are other goals of these ongoing investigations.
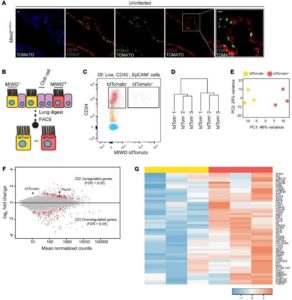
Figure 3: MIWI2-positive airway cells represent a transcriptionally discrete subpopulation of multiciliated cells. (A) Lung sections from Miwi2+/Tom mice immuno-stained for red fluorescent protein (RFP, white), FOXJ1 (green), and CC10 (red). Scale bars: 10 μm. (B) MIWI2+ cells are labeled through the expression of tdTomato. (C) Cytometry plot selected for live, CD45–, EpCAM+ cells from Miwi2+/Tom mouse after elastase lung digest. (D) Unsupervised hierarchical clustering based on whole transcriptome data from MIWI2+ and MIWI2– ciliated cells. (E) Principal component analysis of the data sets analyzed in C. (F) MA plot representation of DESeq output. Red dots indicate genes determined to be significant at FDR q < 0.05. Piwil4 (an alias of Miwi2) and tdTomato are indicated. (G) Heatmap representation of top 50 genes induced in MIWI2-positive versus MIWI2-negative ciliated cells. Values normalized by Z score. Data presented are from n = 3 mice per group.
RESEARCH ETHICS
Over time, I developed an interest in the complex issues associated with the ethical conduct of research and how best to educate and enlighten trainees about these issues. To pursue this interest, I completed a 1 year fellowship in Bioethics at Harvard University (2014) and now lead, in my capacity as Director of RCR, a formalized educational program on research ethics that is directed at pre-doctoral and post-doctoral students throughout Boston University. In addition to the didactic elements of this program, we have several ongoing studies relating to the pedagogy of research ethics and the relationship of research misconduct to paper retractions.
REPRESENTATIVE PUBLICATIONS
-
Chen F, Shao F, Hinds A, Yao S, Ram-hohan S, Norman T, Krishnan R, and Fine, A. Retinoic Acid signaling is essential for airway smooth muscle homeostasis. JCI Insight doi: 0.1172/jci.insight.120398, 2018 (PMID: 30135301).
Drimer-Batca D, Iaccarino J, and Fine A., Status of retraction notices for biomedical publications associated with research misconduct.doi.org/10.1177/1747016118820496, 2019.
Norman T, Gower A, Chen F, and Fine, A. Transcriptional landscape of pulmonary lymphatic endothelial cells during fetal gestation. PLOS ONE.https://doi.org/10.1371/journal.pone.0216795, 2019 (PMID: 31083674).
Wasserman G, Szymaniak AD, Hinds AC, Yamamoto K, Kamata H, Smith NMS, Hilliard KL, Carrieri C, Labadorf AT, Quintin LJ, Ai X, Varelas X, Chen F, Mizgerd JP, Fine A, O’Carroll DO, and Jones MR. Expression of Piwi protein MIWI2 defines a distinct population of multiciliated cells. Journal Clinical Investigation, https://doi.org/10.1172/JCI94639, 2017 (PMID: 28920925).